Neuropathic pain: mechanisms and their clinical implications
BMJ 2014; 348 doi: https://doi.org/10.1136/bmj.f7656 (Published 05 February 2014) Cite this as: BMJ 2014;348:f7656
- Steven P Cohen, professor1, professor of anesthesiology and physical medicine and rehabilitation2,
- Jianren Mao, professor of anesthesia research and vice chair for research3
- 1Departments of Anesthesiology and Critical Care Medicine and Physical Medicine and Rehabilitation, Johns Hopkins School of Medicine, Baltimore, MD 21029, USA
- 2Uniformed Services University of the Health Sciences, Bethesda, MD, USA
- 3Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA
- Correspondence to: S P Cohen scohen40{at}jhmi.edu
Abstract
Neuropathic pain can develop after nerve injury, when deleterious changes occur in injured neurons and along nociceptive and descending modulatory pathways in the central nervous system. The myriad neurotransmitters and other substances involved in the development and maintenance of neuropathic pain also play a part in other neurobiological disorders. This might partly explain the high comorbidity rates for chronic pain, sleep disorders, and psychological conditions such as depression, and why drugs that are effective for one condition may benefit others. Neuropathic pain can be distinguished from non-neuropathic pain by two factors. Firstly, in neuropathic pain there is no transduction (conversion of a nociceptive stimulus into an electrical impulse). Secondly, the prognosis is worse: injury to major nerves is more likely than injury to non-nervous tissue to result in chronic pain. In addition, neuropathic pain tends to be more refractory than non-neuropathic pain to conventional analgesics, such as non-steroidal anti-inflammatory drugs and opioids. However, because of the considerable overlap between neuropathic and nociceptive pain in terms of mechanisms and treatment modalities, it might be more constructive to view these entities as different points on the same continuum. This review focuses on the mechanisms of neuropathic pain, with special emphasis on clinical implications.
Introduction
Pain is a survival mechanism that serves as a warning sign of ongoing or impending tissue damage. According to an Institute of Medicine report released in 2011, one in three Americans experiences chronic pain—more than the total number affected by heart disease, cancer, and diabetes combined.1 In Europe, the prevalence of chronic pain is 25-30%.2 About a fifth of people who report chronic pain are thought to have predominantly neuropathic pain.3 4
Summary points
Neuropathic pain, defined as pain resulting from injury to, or dysfunction of, the somatosensory system, tends to be more refractory to treatment than other forms of pain
As more accurate instruments have been developed to identify neuropathic pain, estimates of its prevalence and socioeconomic impact have increased
The development of neuropathic pain requires a plethora of different mechanisms that extend from the periphery to the central nervous system where they involve the spinal cord, brain, and descending modulation systems
Although conceptually appealing, the mechanism based treatment of pain is challenging to implement
Many drugs shown to be effective in preclinical models of neuropathic pain fail in clinical studies, mostly because animal models tend to emphasize evoked, rather than spontaneous, pain and do not account for the emotional aspects of pain
In view of the large degree of overlap between neuropathic and nociceptive pain in terms of mechanisms and treatment response, many experts view them as different points on a chronic pain continuum, rather than distinct entities
Rationale for mechanism based treatment
One reason for the high prevalence rate of chronic pain, and neuropathic pain in particular, is the absence of effective treatments. Unlike opioids and non-steroidal anti-inflammatory drugs, which form the cornerstone of drug treatment for nociceptive pain, the adjuvants used to treat neuropathic pain tend to have only a modest effect and in a minority of patients. The main reason for this is the inability to target underlying mechanisms precisely; this is why syndromes (such as fibromyalgia), which lack distinct pathophysiological mechanisms, tend to be associated with lower treatment success rates than diseases.5
Definitions
Allodynia: Painful response to a normally innocuous stimulus
Central pain: A subset of neuropathic pain caused by a lesion or disease of the central somatosensory nervous system
Central sensitization: Increased responsiveness of nociceptive neurons in the central nervous system to normal or subthreshold sensory input
Deafferentation pain: Pathological pain condition associated with a partial or complete loss of sensory input from a part of the body after lesions in somatosensory pathways, often as a result of reorganization in the central nervous system. Common examples include phantom limb pain and brachial plexopathy
Descending modulation: The process by which pathways that descend from the brain to the spinal cord modify incoming somatosensory information so that the perception of and reactions to somatosensory stimuli are altered, resulting in increased or decreased pain
Ectopic discharge: Trains of ongoing electrical nerve impulses that occur spontaneously without stimulation or originate at sites other than the normal location (or both). This phenomenon typically occurs after nerve injury
Ephaptic transmission: The phenomenon by which two independent nerves communicate with each other through an artificial synapse, which often develops after injury to the insulating myelin sheath that normally prevents crosstalk between parallel nerves
Hyperalgesia: Increased pain response to a normally painful stimulus
Neuropathic pain: Pain caused by a lesion or disease of the somatosensory nervous system
Neuroplasticity: Changes in neural pathways and synapses that result from bodily injury or changes in behavior, the environment, or neural processes. This is consistent with the concept that the brain is a dynamic organ that constantly changes in response to internal and outside events throughout life
Nociception: The neural responses of encoding and processing noxious stimuli
Nociceptive pain: Pain that arises from the activation of peripheral nerve endings (nociceptors) that respond to noxious stimulation. Nociceptive pain arises from actual or potential damage to non-neural tissue and can be categorized as visceral or somatic
Noxious stimulus: A stimulus that damages or threatens to damage normal tissues
Peripheral sensitization: A lowering of the stimulus (pain) threshold for nociceptor activation and an increased frequency of nerve impulse firing in response to stimulation (hyperexcitability). Peripheral sensitization is often found at the site of tissue damage or inflammation
Sympathetically maintained pain: Pain that is enhanced or maintained by a functional abnormality of the sympathetic nervous system, such as functional sympathetic afferent coupling or increased expression of adrenergic receptors at the peripheral terminals of nociceptive afferent fibers
Windup: Progressive increase in the frequency and magnitude of firing of dorsal horn neurons produced by repetitive activation of C fibers above a critical threshold, leading to a perceived increase in pain intensity
Generally, mechanism based treatments, which target specific pain mechanisms, are superior to disease based or cause based treatments, which target less proximate causes. This may be one reason why so many drugs that are successful in preclinical studies fail in clinical trials.6 As a general rule, painful conditions such as inflammatory arthritis, in which the mechanisms have been clearly identified, have more effective treatments.7 But in clinical practice, elucidating the pain mechanisms responsible for neuropathic symptoms can be difficult. One method for identifying mechanisms and predicting treatment outcomes is the use of intravenous infusion tests, such as intravenous ketamine to predict response to dextromethorphan or other NMDA (N-methyl-D-aspartate) receptor antagonists. However, studies that have evaluated these treatments have been methodologically flawed and usually have reported only modest predictive value.8
In the past decade, several reviews have been written on the mechanisms of neuropathic pain, most of which are directed at neuroscientists. Yet it is essential that clinicians understand the mechanisms too, because such an understanding can steer future research and guide clinical practice.
Search methods
In September 2013, we searched the databases on Medline via PubMed and Ovid, Embase, and CINAHL Plus using the keywords “neuropathic pain”, “sensitization”, “neuroplasticity”, “mechanisms”, “reorganization”, “sympathetically maintained”, “antinociceptive”, and “descending modulation”, with no date restrictions. For individual sections, keywords relating to specific topics and mechanisms were identified from the initial search (for example, “ion channel expression”, “cytokine”, “glial cell”) and searched using the same databases. Additional articles and prime references were obtained by cross referencing all search terms with “review article” and searching through reference lists. We considered animal studies, experimental and clinical trials, and review articles published in English.
Physiology and classification
The generation of pain in response to tissue injury involves four basic elements:
Transduction: a function of nociceptors that converts noxious stimulation to nociceptive signals
Transmission: a process that sends nociceptive signals along nerve fibers from the site of injury to the central nervous system (CNS)
Transformation or plasticity: a mechanism that modulates nociceptive signals at synaptic sites and at the level of the CNS through ascending, descending, or regional facilitation and inhibition
Perception: a key component of the clinical pain experience that integrates cognitive and affective (emotional) responses.
In evolutionary terms the activation of high threshold mechanical nociceptors or other types of specialized nociceptor served a protective role, acting as a warning system for dangerous stimuli. But whereas inflammatory pain is adaptive, evolution has failed to account for our enhanced ability to survive trauma, disease, or iatrogenic trauma intended to prolong or enhance quality of life (such as surgery). In these contexts pain no longer serves a useful function but becomes the disease itself.
Although it is easy to conceptualize pain as a homogeneous entity, this is overly simplistic. In reality there are several different types, each with distinct neurobiological and pathophysiological mechanisms. The most common categorization divides pain into two main types: neuropathic and nociceptive pain (table 1⇓). This distinction is important because it not only reflects the cause of pain but also informs treatment.
Classification of neuropathic and nociceptive pain
Nociceptive pain can be classified as somatic (for example, muscles, joints) or less often visceral (internal organs). Because of the high concentration of nociceptors in somatic tissues, chronic somatic pain is typically well localized and often results from degenerative processes (such as arthritis). By contrast, internal organs are usually unresponsive to classic painful stimuli, such as cutting and burning, but respond to ischemia (for example, angina), inflammation (appendicitis), or occlusion of flow that results in capsular distension (bowel obstruction).
Neuropathic pain is defined as pain resulting from injury to, or dysfunction of, the somatosensory system.9 In neuropathic pain, tissue damage directly affects the nervous system, resulting in the generation of ectopic discharges that bypass transduction.10 One subtype of neuropathic pain is central pain (for example, as a result of spinal cord injury), which manifests as a constellation of signs and symptoms that follows an insult to the CNS as a necessary, but not always sufficient, inciting event. Although many forms of nociceptive pain, and some forms of neuropathic pain, may confer evolutionary benefits, chronic neuropathic pain is always maladaptive.
Compared with previous studies, estimates of the prevalence of neuropathic pain have significantly increased over the past decade since the development of instruments designed to identify such pain.11 Around 15-25% of people with chronic pain are currently thought to have neuropathic pain.3 4 However, the prevalence of neuropathic pain may belie its socioeconomic impact, because studies have found that it is associated with a greater negative impact on quality of life than nociceptive pain.12
Emotional versus physiological aspects
A common misconception is that pain is purely a physiological phenomenon. In fact, “pain” represents a final integrative package, the components of which consist of neurophysiological processes as well as contextual, psychological, and sociocultural factors. This is one reason for the discrepancies between preclinical studies (which measure increased tolerance to painful stimuli in animals (anti-nociception)), clinical studies (which assess efficacy), and clinical practice, which measures effectiveness (table 2⇓). Partly because of these factors and the neurophysiological differences between individuals, the degree of pathology tends to correlate poorly with the intensity of pain for conditions such as back pain.13 To illustrate, conditions such as fibromyalgia have high reported pain scores despite the absence of overt disease.
Comparison of pain in animal models and clinical pain
Secondary order neurons arising in the spinal cord transmit nociceptive input to the thalamus through ascending pathways such as the spinothalamic tract, which functions as a relay station to higher cortical centers. These centers include:
The anterior cingulate cortex, which is involved in anxiety, anticipation of pain, attention to pain, and motor responses
The insular cortex, which may play a role in the sensory discriminative and affective aspects of pain that contribute to the negative emotional responses and behaviors associated with painful stimuli14
The prefrontal cortex, which is important for sensory integration, decision making, memory retrieval, and attention processing in relation to pain15
The primary and secondary somatosensory cortices that localize and interpret noxious stimuli16
The nucleus accumbens, which is involved in placebo analgesia17
The amygdala, hippocampus, and other parts of the limbic system, which are involved in the formation and storage of memories associated with emotional events, affect, arousal, and attention to pain and learning. The limbic system may also be partially responsible for the fear that accompanies pain.18
Because pain is multidimensional experience, it is not surprising that psychosocial factors such as depression, somatization, poor coping skills, social stressors, and negative job satisfaction can predict the development of chronic pain after an acute episode.19 20 In addition, the context in which a painful stimulus occurs affects how we perceive it. This is why an injury that occurs during a football game may be less painful than a similar injury that occurs while walking to school, and why acute pain, which we anticipate will get better, is better tolerated than chronic pain.
Peripheral mechanisms
Peripheral sensitization
Once injury occurs, inflammation and reparatory processes ensue, leading to a hyperexcitable state known as peripheral sensitization. In most patients, this state resolves as healing occurs and inflammation subsides. However, when nociception persists because of repeated stimulation from ongoing injury or disease (for example, in diabetes), the changes in primary afferent neurons may persist.
Several factors can contribute to peripheral sensitization. Inflammatory mediators such as calcitonin gene related peptide and substance P, which are released from nociceptive terminals, increase vascular permeability, leading to localized edema and the escape of the byproducts of injury, such as prostaglandins, bradykinin, growth factors, and cytokines. These substances can sensitize as well as excite nociceptors, resulting in lowered firing thresholds and ectopic discharges. The fact that multiple substances can sensitize nociceptors may partly explain why no drug is universally effective and there is a ceiling effect for antagonists that work at only one receptor (such as non-steroidal anti-inflammatory drugs (NSAIDs)).
Ectopic discharges can give rise to spontaneous pain and may originate from the dorsal root ganglion, other points along an injured nerve, or even uninjured adjacent fibers.21 The process by which adjacent uninjured nerve fibers become excited as a result of non-synaptic “cross talk” is known as ephaptic transmission. Allodynia refers to pain produced by a normally non-painful stimulus, and it may result from decreased stimulation thresholds. Allodynia can be classified as mechanical (pain in response to light touch) or thermal, and it can readily be detected on physical examination. An example is a patient with diabetic neuropathy whose feet are sensitive to putting on socks.
Hyperalgesia refers to exaggerated pain perception as a result of damaged peripheral pain fibers, and it can be categorized as primary or secondary. Primary hyperalgesia occurs in injured tissue as a result of sensitization of peripheral nociceptors (for example, tenderness after a cut), whereas secondary hyperalgesia is seen in adjacent undamaged tissue owing to sensitization within the CNS and can be assessed with a sharp object. In part, this may be caused by ephaptic transmission or the expansion of receptive fields of injured nerves (or both). A clinical example of hyperalgesia might be an amputee who is unable to use a prosthesis because of tenderness overlying the stump. Both allodynia and hyperalgesia are forms of evoked, or stimulus dependent, pain. Although spontaneous neuropathic pain is often more common and distressing than evoked pain in clinical practice, preclinical studies usually measure evoked pain (fig 1⇓).22 It is still not clear whether animals that develop evoked pain incited by models of peripheral nerve injury experience spontaneous pain.

Fig 1 Diagram depicting normally perceived pain, as well as allodynia and hyperalgesia after injury
{kind=link}
Expression of ion channels
One contributor to spontaneous firing of nerve fibers after injury is the increased expression of sodium channels in dorsal root ganglia and around the terminal injury site (neuroma) of injured axons.23 Since this discovery, further preclinical studies have shown that a variety of sodium channels are involved in pain. After nerve injury, the expression of some of these channels increases de novo, the expression of others diminishes, and some translocate into different cellular compartments.24 The proliferation of heterotopic sodium channels, such as Nav1.3, Nav1.7, and Nav1.8, may lower the stimulation threshold and provoke ectopic discharge, resulting in spontaneous pain. In addition, the spread of sodium channels may trigger central sensitization, leading to allodynia. Several adjuvant drugs, such as carbamazepine, act through the blockade of sodium channels. Yet, because none of these drugs is selective for channel subtypes involved in pain, all have low therapeutic indices and many side effects.
Certain types of calcium channels (N-type, T-type, and L-type), and to a lesser extent potassium channels (hyperpolarization activated cyclic nucleotide gated channels), also play a role in neuropathic pain. After nerve injury, the expression of α2δ calcium channels increases in and around the dorsal root ganglia, increasing excitability.25 These voltage gated calcium channels are the primary site of action for gabapentinoids, a first-line treatment for neuropathic pain,26 which have been shown in preclinical studies to reduce hyperalgesia and spontaneous pain (table 3⇓).27
Evidence for pharmacotherapy based on mechanisms of neuropathic pain
Phenotypic switch
Differentiated neurons have different properties from undifferentiated ones, which enable them to perform specific functions (Aδ and C fibers transmit pain). After nerve injury, hundreds of genes that affect nerve function are upregulated or downregulated, and this can affect excitability, as well as transduction and transmission properties. Because gene expression affects cellular characteristics, this can result in a change in the phenotype of the nerve fiber, such that neuromodulators usually expressed in C fibers (such as calcitonin gene related peptide, substance P) are now expressed in other fibers.28 This may theoretically result in stimuli that are usually innocuous being perceived as painful.
Sensory denervation and sprouting of collateral nerve fibers
After injury to a sensory nerve, atrophic changes (wallerian degeneration) cause a decrease in the size of the cell body and the axon diameter, and eventually neuronal death. This leads to a decreased density of intraepidermal nociceptors. Depending on the type of nerve injury, this may cause loss of sensation or, paradoxically, hyperalgesia and increased pain (deafferentation pain).29 Severing the link between a nerve and its end organ also deprives the nerve of nerve growth factor and other neurotrophins, which are essential for growth and maintenance and serve as signaling molecules. One example of deafferentation pain is phantom limb pain after amputation. Although electrodiagnostic studies may be normal in people with a loss of small pain transmitting nerve fibers, a decreased density of C fibers can be seen on skin biopsy. In response to local release of nerve growth factor, collateral sprouting may follow neuronal loss.
Sympathetically maintained pain
Sympathetically maintained pain is pain that is enhanced or maintained by an abnormality in the sympathetic nervous system. Functional coupling between the sympathetic nervous system and somatosensory nerves after nerve injury has been noted since the American civil war. Although the concept of sympathetically maintained pain is most commonly linked to complex regional pain syndrome, the same principles apply to other pain conditions, such as postherpetic neuralgia.8 The interaction between the anatomically distinct autonomic and somatosensory systems is complex but probably includes the expression of α adrenoceptors on primary afferent sensory fibers, sympathetic sprouting into dorsal root ganglia, and impaired oxygenation and nutrition in response to sympathetically mediated vasoconstriction.30 Clinically, sympathetically maintained pain may manifest as temperature or color changes (or both) in an affected extremity, swelling or atrophy, and pain worsened by cold weather or stress, which enhances sympathetic outflow. Among the various diagnostic tests used to detect sympathetically maintained pain, clinical studies have found that sympathetic blocks are more sensitive but less specific than intravenous infusion of phentolamine.31
Spinal mechanisms
An important spinal component of neuropathic pain mechanisms is synaptic plasticity in the form of temporal and spatial summation (increased neuronal responses to repeated noxious stimulation in a time and region dependent manner).32 Other components include expanded receptive fields of nociceptors and second order neurons,33 and increased neuronal excitability of ascending nociceptive pathways that send pain signals to supraspinal regions.34 These neuroplastic changes take place along nociceptive pathways in the spinal cord and in multiple brain regions.35
In the neural circuit, nociceptive signals generated by nerve damage are modulated by supraspinal descending inhibition or facilitation that converges onto dorsal horn neurons (or both).36 At the cellular level, transmission of nociceptive signals within the central nervous system is regulated by cellular and intracellular elements that include37 38:
Ion (Na+, Ca++, K+) channels
Ionotropic and metabotropic receptors such as glutamatergic, GABA (γ-aminobutyric acid)ergic, serotoninergic, adrenergic, neurokinin, and vanilloid receptors
Inflammatory cytokines released from activated glial cells
Nerve growth factors
Intracellular regulators such as protein kinases (for example, protein kinase C) and transcriptional factors (such as nuclear factor-κB).
Spinal glutamatergic regulation
Peripheral nerve injury increases neuronal excitability in the spinal cord by activating excitatory glutamate receptors.39 Nerve injury also induces downregulation of spinal glutamate transporters responsible for maintaining synaptic glutamate homeostasis. Increased regional glutamate availability secondary to loss of glutamate transporters can result in persistent and enhanced activation of both ionotropic (for example, NMDA and AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid)) and metabotropic glutamate receptors (such as metabotropic glutamate receptor 2), leading to lower activation thresholds and increased neuronal excitability and neurotoxicity.40 41
The term “windup” refers to the progressive increase in the frequency and magnitude of firing of dorsal horn neurons produced by repetitive activation of C fibers, a phenomenon that requires glutamatergic NMDA receptor activity. Spinal glutamatergic activity can in turn initiate intracellular signaling cascades, including activation of protein kinase C, that result in long-lasting neuroplastic changes in the spinal cord.42 Similar to the role of central glutamatergic mechanisms in the pathogenesis of other neurological disorders such as epilepsy and Alzheimer’s disease, glutamate receptors are integral to the development of central sensitization, and blockade of both NMDA and non-NMDA receptors has been shown to attenuate neuropathic pain in animal models.43 Because of its primary role in neuroplasticity and excitotoxicity, the NMDA receptor has been implicated in such diverse areas as memory, opioid tolerance, and opioid induced hyperalgesia—the phenomenon whereby opioid use paradoxically increases pain sensitivity.44 In clinical practice, the use of NMDA receptor antagonists to prevent opioid tolerance and hyperalgesia has been disappointing.45 The long-term use of these drugs to treat chronic neuropathic pain has also had mixed results, and their use may be limited by side effects, particularly psychomimetic ones, which seem to increase in proportion to potency. The use of ketamine infusions as a treatment for refractory neuropathic pain has generated intense interest, although studies are limited by methodological flaws and lack of long term follow-up (table 3).46 The rationale behind these infusions is that high doses may “reset” the nervous system back to its pre-injury state, in essence reversing central sensitization.
Glial activation and proinflammatory cytokines
The role of glial activation and cytokines in neuropathic pain has been extensively studied. Proinflammatory cytokines including interleukin 1β (IL-1β), IL-6, and tumor necrosis factor α (TNF-α) are produced peripherally and centrally in response to nerve injury.47 These proinflammatory cytokines play a crucial role in inflammatory responses after nerve injury through intracellular mediators such as protein kinase C and 3′,5′-cAMP.48 Proinflammatory cytokines also play an important role in sensitization of the CNS and may contribute to allodynia, hyperalgesia, and neuroma formation.49 50 51 In animal models, administration of cytokine inhibitors before nerve injury reduces neuropathology and pain-related behaviors.52 53 However, in controlled clinical trials, most of which were performed in patients with radiculopathy, the use of systemic and neuraxial cytokine inhibitors has been largely disappointing.54 55
Glial cells comprise about 70% of the central nervous system and play an important role in maintenance and homeostasis. Microglia are activated within 24 hours of nerve injury, and astrocytes follow shortly thereafter, with activation persisting for up to 12 weeks. Glial cells undergo structural and functional transformation after injury, with astrocytes releasing a host of different pronociceptive factors, such as prostaglandins, excitatory amino acids, and cytokines.56
Microglial cells comprise less than 20% of spinal glial cells under normal conditions but proliferate rapidly at the dorsal root ganglia and spinal cord after nerve injury.56 57 On activation, microglial cells stimulate the complement component of the immune system and release cytokines, chemokines, and cytotoxic substances such as nitric oxide and free radicals.56 58 59 This proinflammatory milieu begins at synaptic sites in the brain stem and the site of nerve injury but spreads to more distant sites. The ensuing release of cytokines from astrocytes and microglia induces an array of cellular responses such as upregulation of glucocorticoid and glutamate receptors, leading to spinal excitation and neuroplastic changes.60 IL-1β also enhances conditioned “fear memory” (conditioned fear related memories associated with behavioral responses) through glucocorticoids,61 suggesting that proinflammatory cytokines may participate in the affective experience of pain. Drugs that modulate microglia, such as minocycline, pentoxifylline, and propentofylline, have shown some efficacy in preclinical models of neuropathic pain but have not proved effective in a clinical context (table 3; fig 2⇓).62
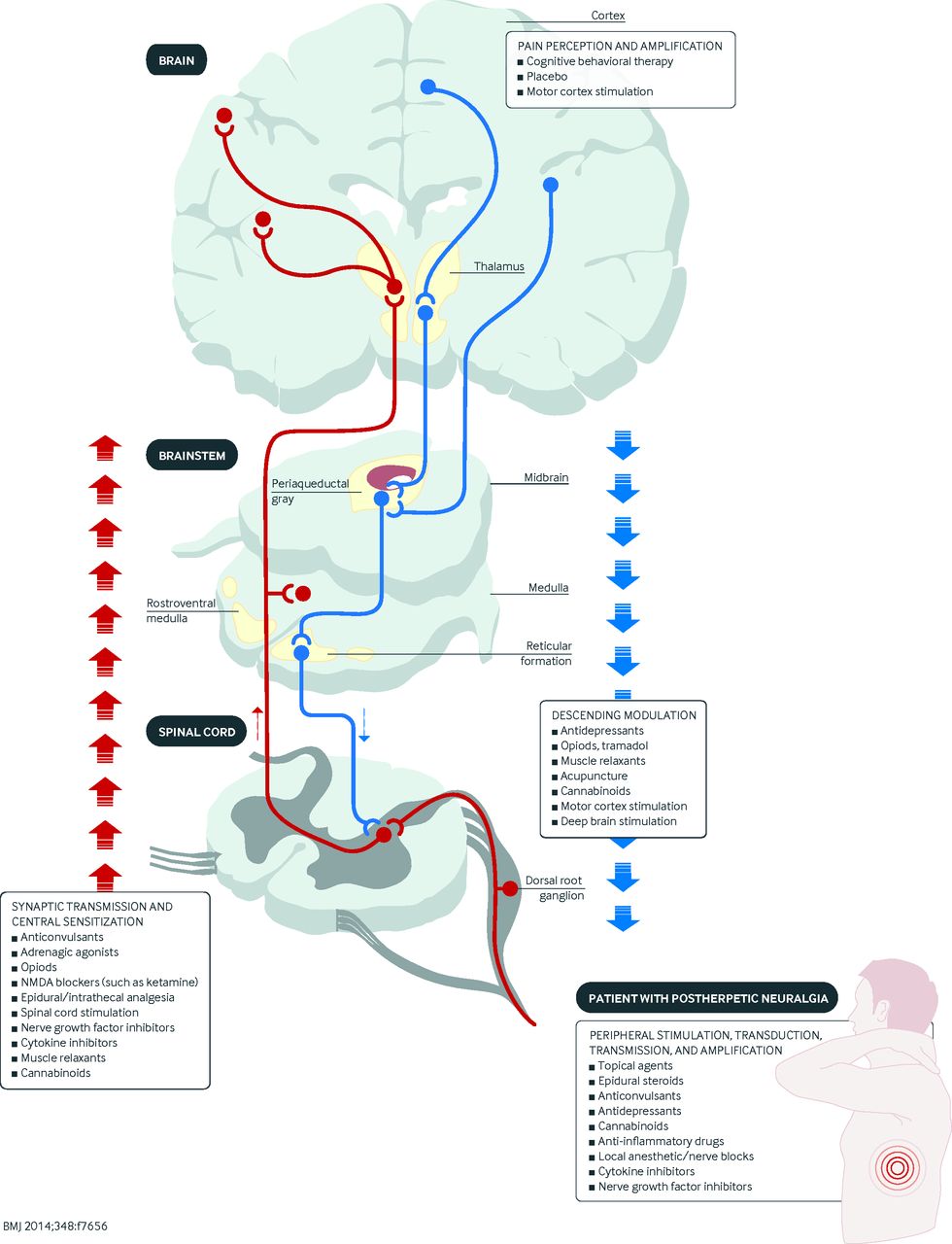
Fig 2 Diagram showing the site(s) of action of various classes of analgesics. NMDA=N-methyl-D-aspartate
{kind=link}
Supraspinal mechanisms
Nociceptive signals can also be altered at supraspinal levels. The brains of patients with chronic pain are different from those without pain, with variations in metabolism and regional concentrations of neurotransmitters occurring in areas such as the thalamus and cingulate cortex. These differences vary according to the type of pain experienced (for example, acute pain or allodynia).63 In patients with neuropathic pain, cortical reorganization occurs after injury, and the extent of the changes seems to correlate with the degree of pain. For example, in upper extremity amputees with phantom limb pain, because of the close proximity of their somatotopic representations, the area of the brain responsible for moving the lips transgresses into the hand movement area of the motor cortex; this phenomenon does not occur in amputees without phantom limb pain.64 The observation that these changes occur after injury suggests that disinhibition may not only be a consequence of nerve injury, but may render patients susceptible to chronic pain.65
Preclinical studies demonstrating changes in gene expression after nerve injury have provided insight into how changes in signal transduction and neuroprotection/apoptosis contribute to neuropathic pain.66 67 Changes that occur in supraspinal regions may explain the strong association between neuropathic pain and mood disorders. Investigators recently found that altered corticotropin releasing factor signaling in the limbic system, an area involved in emotions, may play a role in the development of neuropathic pain.68 Patients with chronic pain have also been shown to have reduced gray matter compared with control patients, and this can be partially reversed by treatment.69
Disinhibition
Spinal cord level
Once a nociceptive stimulus is transmitted to higher cortical centers, a series of events occurs that results in the activation of inhibitory neurons that attenuate pain. At the spinal cord level, there is increased release of GABA and glycine from primary afferent terminals, and enhanced activity in inhibitory GABAergic and glycinergic dorsal horn interneurons. These spinal interneurons synapse with central terminals of primary afferent neurons, thereby reducing their activity, and also regulate activity in ascending secondary order neurons. Spinal inhibitory systems may exert a greater effect on the development of mechanical hyperalgesia than on thermal hyperalgesia.70 71
After nerve injury, a loss of inhibitory currents occurs as a result of dysfunctional GABA production and release mechanisms; impaired intracellular homeostasis from reduced activity of K+Cl− cotransporter or increased activity of Na+K−Cl− cotransporter (or both), leading to increased Cl− levels; and apoptosis of spinal inhibitory interneurons.72 73 Loss of inhibitory control has been shown to provoke tactile allodynia and hyperalgesia,74 and to facilitate structural changes that increase transmission from Aβ fibers that normally transmit non-painful stimuli to nociceptive specific secondary order neurons in the dorsal horn.75
After nerve injury, dorsal root ganglia exhibit decreased expression of µ opioid receptors and secondary spinal neurons become less responsive to opioids.76 By contrast, inflammation may result in an increase in the number and affinity of opioid receptors, thereby enhancing the efficacy of opioids.77 This may explain why patients with chronic neuropathic pain require higher doses of opioids than those with acute and chronic nociceptive pain.78 In preclinical studies, the administration of NMDA receptor antagonists, protein kinase Cγ inhibitors, and GABA-A agonists has been shown to reverse allodynia and hyperalgesia.75 79 Clinical trials provide evidence for a small effect size for ketamine in neuropathic pain, although the high doses given make it difficult to identify the precise mechanism responsible for analgesia.8 46 There is some evidence to support the use of the GABA-B agonist baclofen for trigeminal neuralgia, but most of the evidence in favor of benzodiazepines as analgesics is anecdotal (table 3).79 80
Descending inhibition plays an important role in determining how people experience pain. Recently, it has been shown that descending modulation can be both inhibitory and facilitatory, with conflicting signals often arising from the same regions.81 The balance between inhibition and amplification is dynamic and influenced by context, behavior, emotions, expectations, timing, and pathology. After injury, there is an initial spike mediated by changes in the activation and gene expression of NMDA and AMPA excitatory glutaminergic receptors, and a subsequent decrease in the excitability of neurons in the rostral ventromedial medulla, which lead to facilitation and inhibition, respectively.82 The evolutionary advantage of these changes is that the initial stimulus is reinforced to ensure that it is given priority, but once this occurs the brain seeks to mitigate the consequences.
Expectations and context also play a role in descending modulation. In one randomized study,83 20 healthy subjects were subjected to painful electrical stimulation of the sural nerve after immersion of an arm in cold water. Half the subjects were told that the immersion would decrease the pain, whereas the other half were told that it would exacerbate the pain. Normally, exposure to a spatially distinct noxious stimulus should decrease the response to pain, a concept known as “descending (or diffuse) noxious inhibitory control.” The study found that the analgesia expectancy group experienced a 77% decrease in pain intensity during immersion compared with no significant reduction in pain in the group that anticipated hyperalgesia. Moreover, corresponding changes in activity levels were noted in cortical areas involved in descending inhibition and placebo analgesia.84 These findings agree with other studies that have found that a host of psychosocial factors such as emotions, expectations, and attention affect our intrinsic ability to inhibit pain.84 85 This may explain why positive expectations tend to result in better treatment outcomes and a higher placebo response rate, and why we are less likely to perceive pain when an injury occurs while we are preoccupied (for example, during a sports game rather than at bedtime).86
Supraspinal level
Descending pathways that modulate transmission of nociceptive signals originate in the periaqueductal gray, locus coeruleus, anterior cingulate gyrus, amygdala, and hypothalamus, and are relayed through brainstem nuclei in the periaqueductal gray and medulla to the spinal cord. The inhibitory transmitters involved in these pathways include norepinephrine (noradrenaline), 5-hydroxytryptamine, dopamine, and endogenous opioids. After nerve injury, several processes take place that mitigate the normal pain attenuating pathways. These include a diminution in tonic noradrenergic inhibition and a shift from a predominantly inhibitory role to a facilitative function for descending serotonergic modulation.87 The manifold roles of these neurotransmitters to affect pain, mood, and sleep may partially explain the high comorbidity rates between pain, depression, anxiety, and sleep disturbances.88 Monoamine reuptake inhibitors such as tricyclic antidepressants are not only effective for neuropathic pain and depression but also alleviate anxiety and improve sleep (fig 3⇓).89

Fig 3 Diagram showing the various mechanisms involved in neuropathic pain at different sites in the nociceptive pathway. AMPA=α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; ASIC=acid sensing ion channel; B1/B2=bradykinin receptor 1/2; BDNF=brain derived neurotrophic factor; CCL=chemokine (C-C motif) ligand; CC-R2=CC-chemokine receptor; DAMPs=danger associated molecular patterns; EPR=prostaglandin E2 sensitive receptor; GABA: γ-aminobutyric acid; Glu=glutamate; H1R=histamine receptor; 5-HT=5-hydroxytryptamine; IL=interleukin; KCC=potassium-chloride cotransporter; m-Glu=metabatropic glutamate; NGF=nerve growth factor; NK=neurokinin; NMDA=N-methyl-D-aspartate; PAMPs: pathogen associated molecular patterns; PG=prostaglandin; P2X=purinergic receptor channel; -R=receptor; SP=substance P; TLR=toll-like receptor; TNF=tumor necrosis factor; Trk=tyrosine kinase; TTxR=tetrodotoxin resistant sodium channel; TTxS=tetrodotoxin sensitive sodium channel; VR=vanilloid receptor (transient receptor potential cation channel subfamily V member 1 TRPV-1)
{kind=link}
Neuropathic versus nociceptive pain: different entities or part of the same continuum?
It is generally acknowledged that neuropathic and non-neuropathic pain are distinct entities, but some experts dispute this assertion, considering it part of our natural tendency to categorize things. There are two main factors that distinguish neuropathic pain from nociceptive pain:
Nociceptive pain requires transduction to convert a non-electrical signal (for example, mechanical) to an electrochemical one, whereas neuropathic pain involves direct nerve stimulation
Different prognosis: most people with nociceptive pain (for example, after surgery) recover, but injury to a major nerve (for example, plexopathy or limb amputation) often results in persistent pain.90
Even the requirement for “nerve injury” in neuropathic pain is contentious. After a nociceptive stimulus, we feel pain because microscopic nerve fibers are embedded in the injured tissue. The difference between neuropathic and non-neuropathic pain might therefore be considered one of scope (large v small nerve injury), although many forms of neuropathic pain, such as small fiber neuropathy, also do not involve discrete nerve injury.
Neuroscientists use distinct models for non-neuropathic (for example, Carrageenan) and neuropathic pain, and even different models (>40) of neuropathic pain to reflect myriad causes (for example, chronic constriction injury, spared nerve injury models).91 Yet, the same neurotransmitters, neuropeptides, cytokines, and enzymes are implicated in both types of pain, with a large degree of overlap. NMDA receptor antagonists are often considered to be effective for neuropathic pain only, being intricately involved in the process of central sensitization, but preclinical and clinical studies have shown that they alleviate nociceptive pain too.46 92 93 Similarly, the voltage gated calcium channel subunit α-2δ-1 is upregulated in injured dorsal root ganglion neurons but not in inflammatory pain.25 However, drugs that block these channels, such as gabapentin, are effective in both preclinical models of nociceptive pain94 and in preventing chronic postsurgical pain when given pre-emptively.95 Conversely, drugs widely acknowledged to be effective only for nociceptive pain may also alleviate neuropathic pain. NSAIDs are so widely viewed as being ineffective for neuropathic pain that no major guidelines even mention them in their algorithm.26 But preclinical and clinical studies have demonstrated efficacy for NSAIDs in neuropathic pain states,96 97 and they are commonly prescribed for neuropathic pain (tables 2 and 3).98
It is important to note that ascending spinal pathways, supraspinal regions that process these signals, and descending modulation pathways are essentially the same for neuropathic and non-neuropathic pain. This creates a difference between the taxonomic classification of pain and the functional and practical classification. Considering the large overlap between neuropathic and nociceptive pain, similar to the classification of other neurological disorders (such as tension-type and migraine headaches) that share pathophysiological mechanisms and overlap in their response to treatment,99 the different types of chronic pain might best be viewed as points on the same continuum.
Emerging treatments
It is anticipated that translational pain research will play an important role in understanding pain mechanisms, formulating treatment and research paradigms, and developing new analgesics in the next decade. To facilitate this, several emerging developments must unfold.
Firstly, new animal models should account for the influence of clinical comorbidities such as depression on nociceptive behaviors.100 This will be challenging, because animal models for emotional outcomes tend to be less studied than those for physiological parameters.
Secondly, behavioral assessment tools should be capable of measuring the various dimensions of pain experiences, such as the use of conditional place preference or aversion (forms of pavlovian conditioning used to measure the motivational effects of positive and negative experiences) and behavioral coding in preclinical studies.101 102 For example, because the relief of pain is a reward in itself, analgesic agents that are not rewarding in the absence of pain should become rewarding only in the presence of pain.
Thirdly, the association between brain reorganization seen on advanced imaging and the chronicity of pain should be further explored, with emphasis on how changes on imaging relate to pain behaviors and response to treatment.103
Lastly, the identification of biomarkers and the genotyping or phenotyping of pain characteristics may provide tools that enable us to understand better the heterogeneity of clinical pain and formulate individualized treatment regimens.104 105
These research advances, together with the development of newer drugs tailored to individual patients and specific pain mechanisms, will probably improve the treatment of neuropathic pain in the coming years.
Conclusions
Injury to the peripheral or central nervous system results in maladaptive changes in neurons along the nociceptive pathway that can cause neuropathic pain. Unlike acute pain, chronic neuropathic pain confers no individual or evolutionary advantage and is often considered to be a disease in itself. The myriad mechanisms involved in neuropathic pain overlap considerably with non-neuropathic pain and other neurological conditions. Although treatment based on the mechanism(s) of pain is widely accepted to be theoretically better than treatment based on the cause of pain, or empirical treatment, this paradigm can be difficult to implement in clinical practice. The multitude of different mechanisms, and the affective-motivational component of chronic pain that distinguishes “human pain” from nociception tested in preclinical pain models, make neuropathic pain notoriously refractory to treatment. This in turn has resulted in chronic pain being considered not only a medical problem but also a socioeconomic concern that requires urgent attention.
Key research questions
Is it possible to devise a valid animal model that accounts for the “affective-motivational” (emotional) aspect of pain as well as the “sensory-discriminative” (physio-anatomical) aspect?
Are there any measures that can be taken before (pre-emptive analgesia) or during the early phase after nerve injury that can prevent the transition to chronic neuropathic pain?
Is neuropathic pain represented differently in the brain than other chronic pain conditions?
Can we develop better animal models to reflect spontaneous pain, rather than those that emphasize stimulus dependent pain (for example, allodynia), which may be less relevant in clinical practice?
Although the concept of mechanism based pain treatment is intellectually enticing, can this be routinely incorporated into clinical practice?
Notes
Cite this as: BMJ 2014;348:f7656
Footnotes
Thanks to Srinivasa Raja and Tony Yaksh for their help.
Contributors: SPC conceived, designed, partly wrote, and reviewed the article and tables, and helped with the figures. JM wrote part of the article and tables and critically reviewed the article.
Funding: Funded in part by the Centers for Rehabilitation Sciences Research, Uniformed Services University of the Health Sciences, Bethesda, MD, USA.
Competing interests: We have read and understood the BMJ Group policy on declaration of interests and declare the following interests: None.
The opinions or assertions contained herein are the private views of the authors and must not be construed as official or as reflecting the views of the US Department of the Army or the Department of Defense.
Provenance and peer review: Commissioned; externally peer reviewed.
References
Log in
Log in using your username and password
Log in through your institution
Subscribe from £173 *
Subscribe and get access to all BMJ articles, and much more.
* For online subscription
Access this article for 1 day for:
£38 / $45 / €42 (excludes VAT)
You can download a PDF version for your personal record.