Homocysteine and cardiovascular disease: evidence on causality from a meta-analysis
BMJ 2002; 325 doi: https://doi.org/10.1136/bmj.325.7374.1202 (Published 23 November 2002) Cite this as: BMJ 2002;325:1202
- David S Wald, specialist registrar in cardiology (davidwald{at}hotmail.com)a,
- Malcolm Law, professorb,
- Joan K Morris, senior lecturerb
- a Department of Cardiology, Southampton General Hospital, Southampton SO16 6YD
- b Wolfson Institute of Preventive Medicine, Barts and the London School of Medicine and Dentistry, London EC1M 6BQ
- Correspondence to D S Wald
- Accepted 12 August 2002
Abstract
Objective: To assess whether the association of serum homocysteine concentration with ischaemic heart disease, deep vein thrombosis and pulmonary embolism, and stroke is causal and, if so, to quantify the effect of homocysteine reduction in preventing them.
Design: Meta-analyses of the above three diseases using (a) 72 studies in which the prevalence of a mutation in the MTHFR gene (which increases homocysteine) was determined in cases (n=16 849) and controls, and (b) 20 prospective studies (3820 participants) of serum homocysteine and disease risk.
Main outcome measures: Odds ratios of the three diseases for a 5 μmol/l increase in serum homocysteine concentration.
Results: There were significant associations between homocysteine and the three diseases. The odds ratios for a 5 μmol/l increase in serum homocysteine were, for ischaemic heart disease, 1.42 (95% confidence interval 1.11 to 1.84) in the genetic studies and 1.32 (1.19 to 1.45) in the prospective studies; for deep vein thrombosis with or without pulmonary embolism, 1.60 (1.15 to 2.22) in the genetic studies (there were no prospective studies); and, for stroke, 1.65 (0.66 to 4.13) in the genetic studies and 1.59 (1.29 to 1.96) in the prospective studies.
Conclusions: The genetic studies and the prospective studies do not share the same potential sources of error, but both yield similar highly significant results—strong evidence that the association between homocysteine and cardiovascular disease is causal. On this basis, lowering homocysteine concentrations by 3 μmol/l from current levels (achievable by increasing folic acid intake) would reduce the risk of ischaemic heart disease by 16% (11% to 20%), deep vein thrombosis by 25% (8% to 38%), and stroke by 24% (15% to 33%).
What is already known on this topic
What is already known on this topic There is an association between serum homocysteine concentration and cardiovascular disease, but it is not known whether the association is causal
A common single gene mutation that reduces the activity of an enzyme involved in folate metabolism (MTHFR) is associated with a moderate (20%) increase in serum homocysteine
What this study adds
What this study adds A meta-analysis of MTHFR studies shows a significantly higher risk of both ischaemic heart disease and deep vein thrombosis (with or without pulmonary embolism) in people with the MTHFR mutation
A meta-analysis of prospective studies shows a significant association between homocysteine concentration and ischaemic heart disease similar in size to that expected from the results of the MTHFR studies and a significant association with stroke
The MTHFR studies and the prospective studies do not share the same potential sources of error but both yield similar results—strong evidence that the association between homocysteine and cardiovascular disease is causal
On this basis a decrease in serum homocysteine of 3 μmol/l (achievable by daily intake of about 0.8 mg folic acid) should reduce the risk of ischaemic heart disease by 16%, deep vein thrombosis by 25%, and stroke by 24%
Introduction
The serum concentration of the amino acid homocysteine is positively associated with the risk of ischaemic heart disease, deep vein thrombosis and pulmonary embolism, and stroke. 1 2 There is uncertainty over whether these associations are causal. 3 4 Resolving the question of causality is important because serum homocysteine can be lowered by the B vitamin folic acid, 5 6 raising the prospect of a simple and safe means of prevention.
Three rare autosomal recessive disorders (the homocystinurias), in which homozygotes lack one of the three principal enzymes in homocysteine metabolism, cause extremely high serum homocysteine levels (>100μmol/l) and a high risk of premature cardiovascular disease (affecting half of homozygotes by the age of 307). The biochemical change that is common to all three disorders is a high serum homocysteine concentration (no other substance is consistently high or low), 8 9 indicating that high homocysteine levels cause cardiovascular disease.
Whether moderate increases in serum homocysteine cause cardiovascular disease has been the subject of debate. Moderate increases occur as a result of a mutation in the gene coding for the enzyme methylenetetrahydrofolate reductase (MTHFR) in which cytosine is replaced by thymidine (C→T) at base position 677 of the gene. This variant of the enzyme has reduced activity,10 resulting in an elevation of serum homocysteine concentrations of about 20%.11 The presence of the mutation offers a natural experiment in which the population have, in effect, been randomly allocated to one group with higher homocysteine concentrations or one with a lower level. There is no reason to expect that the two groups differ in any systematic way other than in their serum homocysteine. The C→T mutation is surprisingly common, with about 10% of people in the population being homozygous affected (TT), 47% homozygous unaffected (CC), and 43% heterozygotes (CT). However, because the increase in homocysteine is relatively small and the impact of the mutation varies according to the folate intake in the population, studies comparing the risk of cardiovascular disease in people with and without the TT mutation need large numbers to be able to show an effect if one were present. Previous meta-analyses have had too few studies available to do this.11–13
We present a new meta-analysis of the MTHFR studies (including data from an additional 34 studies since the previous meta-analyses), which has the statistical power to show an effect. We compare these results with those of a meta-analysis of prospective studies of serum homocysteine and disease events. We assess the evidence for causality between serum homocysteine and ischaemic heart disease, deep vein thrombosis with or without pulmonary embolism, and stroke and estimate the size of the associations with the three diseases.
Methods
Identification of studies
We sought two types of study on the association between serum homocysteine and ischaemic heart disease, deep vein thrombosis, or stroke:
Studies reporting the prevalence of the genetic variant of the MTHFR enzyme(homozygous (TT) and heterozygous (CT) compared with “wild type” (CC)) in cardiovascular disease cases and controls.w1-w94Some of these studies also reported the associated differences in serum homocysteine
rospective (cohort) studies of serum homocysteine and disease events.w95-w120 Serum was collected at baseline, and homocysteine concentration measured either then on all subjects or later on stored samples from those who subsequently developed disease events and from matched controls (nested case-control design).
To identify studies, we searched databases (Medline, CINAHL, Embase, the Cochrane Library, PsycINFO, and ClinPSYC) in any language up to October 2001. Subjectheadings were homocysteine, homocystine, methylenetetrahydrofolate reductase or MTHFR andischaemic heart disease, coronary artery disease, myocardial infarction, cerebrovascular disease, stroke, deep vein thrombosis, pulmonary embolism, or thromboembolic disease. We included studies that measured fasting or non-fasting serum or plasma homocysteine concentration using any standard laboratory method but excluded studies reporting only homocysteine measured after methionine loading. We excluded studies of children (aged <18 years) and of haemodialysis or renal transplant patients. In the MTHFR studies we restricted our analysis to studies in which the genetic variant was confirmed by DNA testing.
The primary search generated 171 potentially relevant articles, of which 96 met the above criteria. We also scanned the citations lists of these 96 studies and of review articles, yielding 24 extra studies to make 120 studies in total.
Definition of outcomes
In the MTHFR studies patients with ischaemic heart disease had myocardial infarction or angiographically confirmed coronary artery occlusion (>50% of the luminal diameter). Cases of deep vein thrombosis cases included patients with or without pulmonary embolism. Stroke cases were cerebral infarction or cerebral haemorrhage identified either clinically or by computed tomography or magnetic resonance imaging. The control groups were from the general population, except for some ischaemic heart disease studies, when they were patients who underwent coronary angiography with normal coronary arteries. In cohort studies the end points were death from ischaemic heart disease or non-fatal myocardial infarction, or fatal stroke or non-fatal cerebral infarction or haemorrhage. Data for analysis were extracted independently by two of the authors, and the data sets cross checked.
Statistical methods
For each MTHFR study we determined the odds ratio of being homozygous (TT), and in a separate analysis heterozygous (CT), for the mutant allele compared with being homozygous for the wild type allele (CC) in cases and controls.
In the prospective studies we estimated odds ratios for a 5 μmol/l increase in serum homocysteine (a standard reference increment). In studies that did not report this directly we estimated it as follows. In studies reporting an odds ratio for a specified proportional difference in serum homocysteine we took the homocysteine values 2.5 μmol/l above and below the study mean and, from the proportional difference between these two values, calculated the odds ratio corresponding to this 5 μmol/l difference in homocysteine. In studies reporting odds ratios in subgroups defined by ranked homocysteine concentration we fitted a logistic regression line to the data.
We used a random effects model to derive summary odds ratios from combinations of studies to allow for any heterogeneity across studies. In the prospective studies we adjusted the summary odds ratios for regression dilution bias14; these arose because the serum homocysteine values in each person were from single measurements, which are unrepresentative of a person's long term average value over the period of follow up. We corrected for regression dilution bias by raising the summary odds ratio to the power of the ratio of total (between-person plus within-person) variance to between-person variance. With serum homocysteine this ratio increases over time15 (in addition to random short term fluctuations in serum homocysteine within a person6 there may be longer term variations from a variable rate of increase in homocysteine with age). We used a published estimate of 1.33 for the correction factor in the prospective studies (5 year average interval between blood collection and cardiovascular disease events).15
Results
Ischaemic heart disease
Studies of MTHFR mutation
Fig 1 shows the results of the 46 MTHFR studies of ischaemic heart disease (12 193 cases in total, mean age at time of event 55 years). The odds ratios of homozygotes for the mutant allele (TT) compared with wild type homozygotes (CC) are shown, with the studies ranked in order of increasing effect. There was a wide range of estimates, but the summary odds ratio was 1.21 (95% confidence interval 1.06 to 1.39; P=0.006), indicating that risk was on average 21% higher in TT homozygotes than in CC homozygotes. The summary odds ratio for the heterozygous state (CT v CC) was 1.06 (0.99 to 1.13; P=0.09).

Results of published studies of association between MTHFR mutation and ischaemic heart disease: values are odds ratios (95% confidence intervals) for homozygotes for mutant allele (TT) v wild type (CC)
{kind=link}
From the 33 studies that provided relevant data, the mean difference in serum homocysteine concentration between the TT and CC genotypes was 2.7 μmol/l (2.1 to 3.4 μmol/l) and that between the CT and CC genotypes was 0.29 μmol/l (0.20 to 0.39 μmol/l) (table 1). The summary odds ratio of 1.21 for TT v CC is equivalent to an odds ratio of 1.42 (1.11 to 1.84) for the standard 5 μmol/l increase in serum homocysteine (calculated by raising 1.21 to the power of 5/2.7).
Studies reporting serum homocysteine concentrations (μmol/l) according to MTHFR genotype (homozygotes for the mutant allele (TT), heterozygotes (CT), and wild type homozygotes (CC))
Combining the results from 12 of the MTHFR studies in which other cardiovascular risk factors were measuredw7 w9 w19 w20 w37 w38 w42 w46 w86 w89 w93 w94 showed thatTT and CC homozygotes had similar serum cholesterol concentration, blood pressure, smoking prevalence, and body mass index, with no significant differences. The upper confidence intervals of these risk factors' associations with TT homozygotes were 0.02 mmol/l, 3 mm Hg, 6%, and 0.9 kg/m2 higher respectively; applying these to the quantitative association of each risk factor with ischaemic heart disease16 showed that even if there were confounding by another risk factor it could at most account for half the 21% excess risk seen in TT homozygotes.
Prospective studies and cardiovascular disease
Fig 2 (upper part) shows the results of 16 prospective studies on ischaemic heart disease (3144 events, mean age at event 62 years).w95-w120 The odds ratios are adjusted for age, sex, smoking, blood pressure, and serum cholesterol concentration in all the studies except one, which is adjusted for age and sex only.w106 The summary odds ratio was 1.23 (1.14 to 1.32) for a 5 μmol/l increase in serum homocysteine concentration, or 1.32 (1.19 to 1.45) adjusted for regression dilution bias. There was a suggestion of a trend across the prospective studies for a decreasing odds ratio with increasing age at the time of the event (P=0.07), as has been observed for other cardiovascular risk factors (smoking, serum cholesterol, and blood pressure).

Results of prospective studies of serum homocysteine concentration and ischaemic heart disease and stroke: values are odds ratios (95% confidence intervals) for a 5 μmol/l increase in serum homocysteine, adjusted for age, sex, smoking, cholesterol concentration, and blood pressure (except in one study, adjusted for age and sex alonew106) but not for regression dilution bias
{kind=link}
Deep vein thrombosis
Fig 3 shows the results of the 26 MTHFR studies of deep vein thrombosis (3439 cases, mean age at event 44 years). The summary odds ratio for homozygotes for the mutant allele (TT) compared with wild type homozygotes (CC) was 1.29 (1.08 to 1.54; P=0.007), equivalent to an odds ratio of 1.60 (1.15 to 2.22) for a 5 μmol/l increase in serum homocysteine concentration. The summary odds ratio associated with the heterozygous state (CT v CC) was 1.05 (0.94 to 1.19; P=0.41).
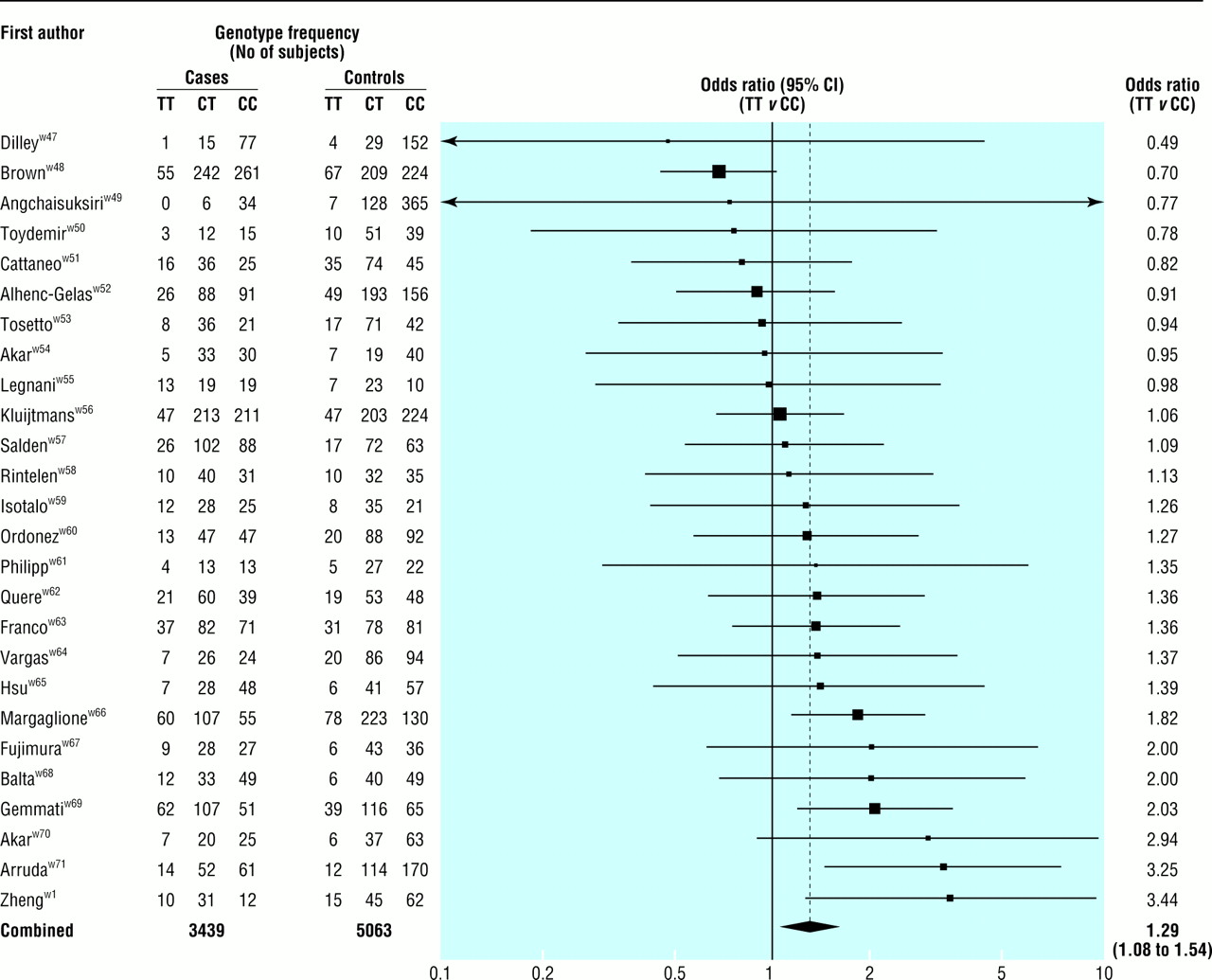
Results of published studies of association between the MTHFR mutation and deep vein thrombosis: values are odds ratios (95% confidence intervals) for homozygotes for mutant allele (TT) v wild type (CC)
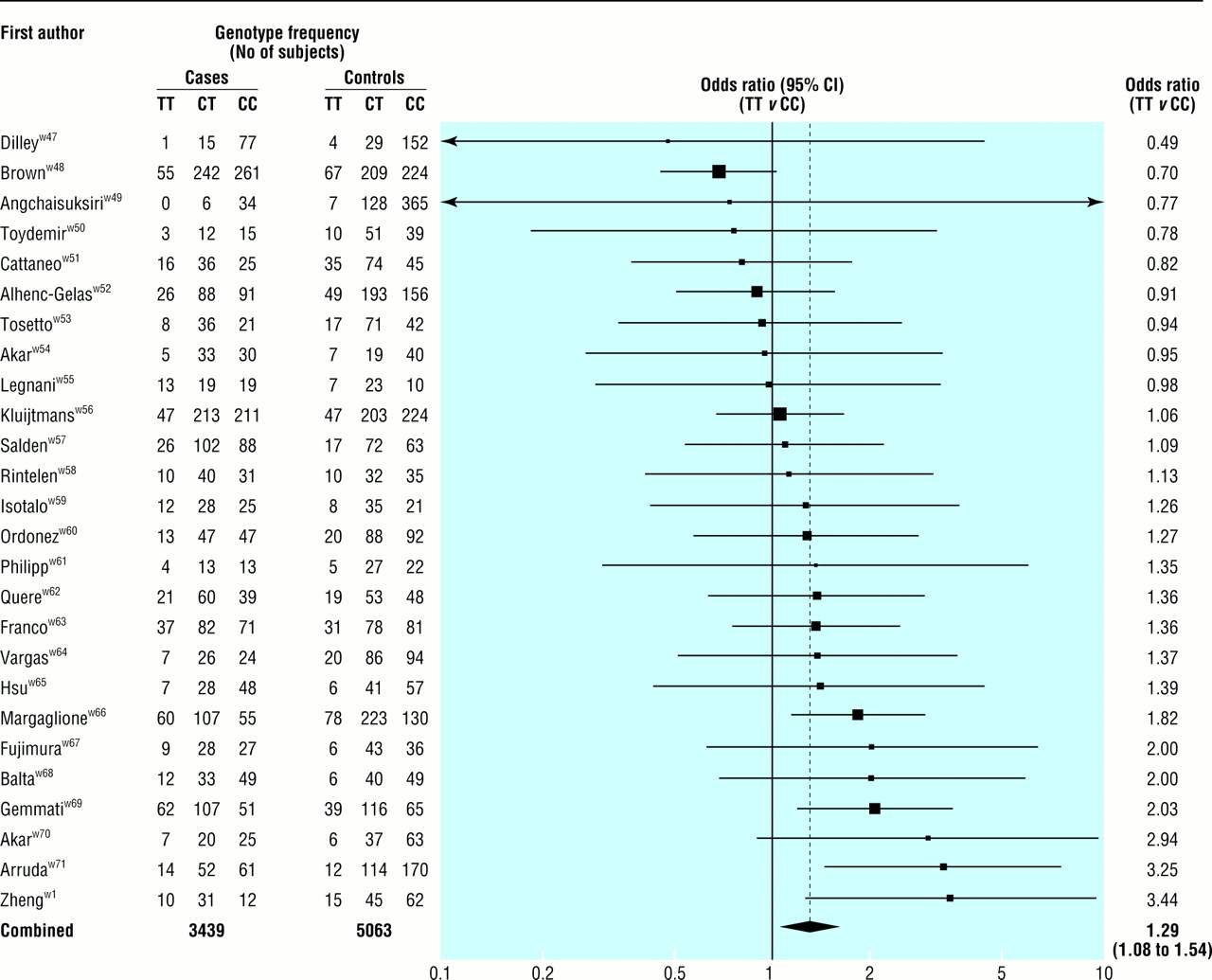{kind=link}
There were no published prospective studies.
Stroke
The seven MTHFR studies of stroke (1217 cases, mean age at event 63 years) yielded relatively few data, so the confidence interval for the summary result was wide: the odds ratio for homozygotes for the mutation (TT) compared with wild type homozygotes was 1.31 (0.80 to 2.15), equivalent to an odds ratio of 1.65 (0.66 to 4.13) for a 5 μmol/l increase in serum homocysteine. The summary odds ratio associated with the heterozygous state (CT v CC) was 1.15 (0.93 to 1.42).
Fig 2 (lower part) shows the results of the eight prospective studies (676 events, mean age at event 70). The results are adjusted for age, sex, smoking, blood pressure, and serum cholesterol in all the studies. The summary odds ratio for a serum homocysteine increase of 5 μmol/l was 1.42 (1.21 to 1.66), or 1.59 (1.29 to 1.96) adjusted for regression dilution bias.
Studies excluded
Four prospective studies could not be included in the analysis. Two reported only on all cardiovascular diseases in combination; these both showed positive associations between homocysteine concentration and mixed vascular disease.w118 w119 Two studies reported odds ratios only relative to a specified homocysteine cut-off value.w92 w116 We could not accurately calculate an odds ratio for a 5 μmol/l increase in homocysteine from this, but estimating a value for these two studies and including them in the analysis had negligible effect (the odds ratio for ischaemic heart disease did not change, that for stroke changed from 1.59 to 1.56).
Discussion
The MTHFR studies show highly significant associations between serum homocysteine concentration and ischaemic heart disease and deep vein thrombosis. Previous meta-analyses of the MTHFR studies failed to show an effect because they lacked statistical power, but their confidence intervals were consistent with the significant estimates we present here. 11–13 17 Our analyses combine data from an extra 23 studies on ischaemic heart disease and an extra 11 studies on deep vein thrombosis since the largest previous meta-analyses. 12 17
The MTHFR studies on ischaemic heart disease and stroke showed significant heterogeneity (greater variation between study results than would be expected through chance). This is not surprising since the expression of the mutation is likely to depend on environmental factors, particularly dietary folate,11 which reduces homocysteine levels. Thus, in MTHFR studies carried out in areas where folate intake is low one would expect to find a larger difference between TT and CC homozygotes in rates of heart disease but also a larger difference in homocysteine concentrations. Heterogeneity, therefore, should not invalidate the analysis provided the results are expressed in terms of the associated differences in homocysteine concentrations.
The prospective studies show a highly significant association between serum homocysteine concentration and both ischaemic heart disease and stroke. The absence of significant heterogeneity across the 16 studies (P=0.12) does not support the view 3 4 9 that these studies divide into two groups, one with clearly positive results and one with clearly negative results (this can also be seen in fig 2).
Interpretation of evidence for causality
The results of the MTHFR and the prospective studies can be explained in one of two ways—a direct (or causal) explanation or an indirect (non-causal) explanation. An indirect explanation would depend on the MTHFR and prospective studies both showing associations with homocysteine through confounding. In the MTHFR studies the difference in homocysteine concentration arises from a single gene mutation effectively allocated at random. There is therefore no basis for expecting that people with the mutant gene would systematically differ from those without it in other cardiovascular risk factors (smoking, blood pressure, serum cholesterol concentration), and the data show that they do not. Genetic confounding is theoretically possible, whereby a gene linked to the MTHFR gene controls some unknown risk factor and coincidentally increases serum homocysteine concentration.
In the prospective studies confounding from smoking, blood pressure, and serum cholesterol was allowed for; if there were any remaining confounding it would also be from an unknown factor associated with both serum homocysteine and cardiovascular disease. Importantly, the genetic linkage that one would have to postulate to explain the association with cardiovascular disease in the MTHFR studies could not account for the association in the prospective studies—because the increase in risk of about a quarter with the MTHFR variant is so much smaller than the twofold difference in risk (10th to 90th centile) in the prospective studies. The indirect explanation relies on a complex and improbable series of different associations that coincidentally yield similar cardiovascular disease risks for a given difference in serum homocysteine concentration. The direct (causal) explanation is plausible and much simpler.
Substantial publication bias is unlikely because many of the individual study results were not statistically significant (those in which the confidence intervals cross 1.0 in figs 1 and 3), and many of these were interpreted by their authors as being negative. A standard statistical assessment of publication bias (the regression asymmetry test18) showed no basis for concern in either the ischaemic heart disease studies (P=0.55) or the deep vein thrombosis studies (P=0.43).
Evidence of risk reduction
A placebo controlled randomised trial of treatment with B vitamins (folic acid, B-6, and B-12) to lower serum homocysteine concentration in patients with ischaemic heart disease has shown a rapid reduction of risk.19 There were 13 major cardiac events (coronary death, non-fatal myocardial infarction, or revascularisation procedure) over six months in 102 vitamin treated patients and 23 events in 94 placebo treated patients (P=0.04).19 Studies of patients with homocystinuria treated with B vitamins have also shown reduction in risk. In one study treatment with B vitamin led to two vascular events when 30 would have been expected (from previous observation in untreated patients), and in another study there were two events when 21 would have been expected. 7 20
Summary results from the MTHFR studies and the prospective studies on risk of ischaemic heart disease, deep vein thrombosis with or without pulmonary embolism, and stroke associated with serum homocysteine concentration
Table 2 summarises the odds ratios of ischaemic heart disease, deep vein thrombosis, and stroke for the standard 5 μmol/l increase in homocysteine concentration and shows combined odds ratios from the two types of study. For ischaemic heart disease this was 1.33 (95% confidence interval 1.22 to 1.46), and for stroke it was 1.59 (1.30 to 1.95). Table 2 also converts the odds ratios for a 5 μmol/l increase in homocysteine concentration into odds ratios for a 3 μmol/l decrease in homocysteine (the maximal effect of folic acid, achieved with a daily dose of about 0.8 mg). 5 6 On the basis that the association is causal and reversible, we estimate that folic acid could reduce the risk of ischaemic heart disease by 16%, deep vein thrombosis by 25%, and stroke by 24%. The folic acid could be taken as tablets by high risk patients, and possibly supplied to the general public through food fortification or a combination of both.
Conclusion
Our results strengthen the evidence that a raised serum homocysteine concentration is a cause of cardiovascular disease. Our conclusion that it is a cause rests on the following observations:
The genetic (MTHFR) studies show a moderate increase in risk for a moderate increase in serum homocysteine; genetic linkage to some unknown risk factor might be the explanation although no such linkage is known
The prospective studies show an association between serum homocysteine and cardiovascular disease after allowance for confounding
These two types of study are susceptible to different sources of error but show quantitatively similar associations, a result that is unlikely to have occurred through different potential sources of confounding acting independently
The homocystinurias cause high serum homocysteine levels and high risks of premature cardiovascular disease
Lowering serum homocysteine reduced risk, both in a randomised trial in patients with heart disease and in patients with homocystinuria.
In the light of these five observations we could not have concluded otherwise.
Acknowledgments
We thank Nicholas Wald for his suggestions throughout this project and for his comments on the manuscript. DSW also holds an honorary research fellowship at the Wolfson Institute of Preventive Medicine, St Bartholomew's and the Royal London School of Medicine and Dentistry. At the time of our checking the proofs of our paper, two other meta-analyses were published in relation to ischaemic heart disease and stroke, yielding results consistent with our own. 21 22
Contributors: DW, ML and JM were involved in the analysis and interpretation of the data and in writing the manuscript. DW is guarantor for the study.
Footnotes
-
Funding None.
-
Competing interests None declared.
-
 The references used in the meta-analysis appear on bmj.com
The references used in the meta-analysis appear on bmj.com